
|
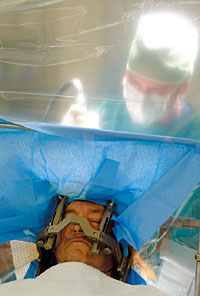 A.J. Anderson, a manager for the Tennes- see Park System for 35 years, retired last July because of the worsening symp- toms of his Parkinson's disease. This fall, neurosurgeon Robert Gross and his team implanted an electrode the size of a thumb- nail deep in Anderson's brain to help him control his shaking hands and head. Anderson is one of thousands of clinical trial volunteers who come to Emory each year to test innovative therapies for a wide variety of diseases and conditions.  |
Jesse Gelsinger was sick, but he was able to manage his symptoms caused by a genetic liver defect. Still, the 18-year-old ultimately wanted to help researchers find a cure -- a common motivation for clinical trial volunteers. In September 1999, Gelsinger was injected with corrective gene material; four days later he was dead. At first, it looked like an unfortunate and rare immune reaction, but the FDA soon shut down the University of Pennsylvania research program after an agency investigation uncovered multiple lapses, some of which may have contributed to Gelsinger's death. Worse still, the scientist leading the work had founded a biotechnology company that might have profited from the research, prompting a conflict-of-interest investigation. Then, a Harvard-affiliated hospital quietly suspended a gene therapy experiment after three of its first six patients died. The researcher had not reported the deaths, as required, to the National Institutes of Health (NIH). The Harvard deaths were among the hundreds of serious "adverse events" in gene therapy that swamped the NIH after Gelsinger's death, the Washington Post reported in January 2000, after obtaining a Freedom of Information Act request. Federal rules demand that such reports be filed immediately as problems arise. But 652 of the 691 reports submitted after Gelsinger died had never before been seen by the NIH; that means less than 6% were filed on time. Most researchers said they didn't know they were supposed to contact the NIH if serious illness, or even death, occur in gene therapy trials. Even before problems surfaced at Penn and Hopkins, the then-called Office for Protection from Research Risks (OPRR), the government's watchdog agency, swept into Duke University Medical Center and stopped all noncritical research for a week. The OPRR said Duke's institutional review board (IRB) was understaffed and overburdened, had failed to correct organizational and administrative problems, and could not ensure that patients were being protected. It was the first time a top-tier medical center had received such a suspension, and it was followed by a clear warning from the Department of Health and Human Services that IRB systems nationwide responsible for safeguarding human subjects were in danger of a meltdown. The federal government's clarion call was heard in Atlanta, and vows were made that Emory would not be the next cited for lax protection of the volunteers entrusted to its care. Now, two years later, Robert Rich, the School of Medicine's executive associate dean for research and strategic initiatives, says Emory meets or exceeds federal regulations. "We've got a process we take considerable pride in." Nevertheless, Emory is not, in any way, complacent and is constantly striving to improve its clinical trials and research process, Rich emphasizes. Hopkins had also reviewed its procedures a year before Roche entered the lung study and had found them solid. "This is an extraordinarily challenging environment," says Rich. "Human research is always difficult, and there are always risks, but research is critical to advancing medicine." On top of that balancing act, research at Emory is rapidly expanding, now up to more than 2,000 active protocols, many of which are clinical trials that depend on patient participation. The number of IRB submissions to Emory has increased 23% in four years, with an accompanying 50% increase in paperwork. With 2,200 active clinical trials, Hopkins itself is not handling that much more than Emory. Research funding is also skyrocketing at Emory University as a whole, up 58% in the past five years to almost $248 million in federal and corporate funding for 2001, 94% for the health sciences. With so much work and so little room for error, can deaths like Ellen Roche's or Jesse Gelsinger's happen at Emory? Jim Keller, chair of Emory's IRB, has lately been assured that many safeguards are in place. But he sees it as his job to be hypervigilant against the possibility that something could happen here. |
  Radiation oncologist Jim Keller and mem- bers of Emory's institutional review board carry heavy responsibility as "guardians" of human subjects. |
 xcept for the white coats and bleeping pagers, it could be a gathering of English faculty poring over the meaning of an obscure text. "'Experimental breath condensate' and 'organic markers' just have to go," says a physician, one of many clustered around a conference table. The committee recommends 8th- to 10th-grade reading levels for most consent forms. For example, when possible, words like "equivalent" are changed to "equal," or better yet, "same." "Adverse" is changed to "negative" or "bad" or "harmful." xcept for the white coats and bleeping pagers, it could be a gathering of English faculty poring over the meaning of an obscure text. "'Experimental breath condensate' and 'organic markers' just have to go," says a physician, one of many clustered around a conference table. The committee recommends 8th- to 10th-grade reading levels for most consent forms. For example, when possible, words like "equivalent" are changed to "equal," or better yet, "same." "Adverse" is changed to "negative" or "bad" or "harmful."
Throughout the meeting, the 16-member IRB committee, which includes faculty, investigators, nonscientists, and community representatives, pull on and off the different "hats" they figuratively wear. Now they are civil rights lawyers, debating whether asking blind patients to sign a consent form constitutes discrimination (see "Sign on the dotted line" sidebar). As ethicists, they question whether paying patients several hundred dollars to participate in a particular clinical trial is a subtle form of coercion. The graphic artist in them suggests that formatting problems need to be corrected and that no words should be underlined. Their medical training questions how any research that involves surgery can be said to be without risk. And so it goes, every Wednesday, for hours at a time in room 307-N, Building A on the Briarcliff Campus. Five separate IRB committees - each meeting once a month - gather to judge requests to enroll people as subjects in Emory studies. The research could be as simple as an anonymous survey or a minor blood draw or as groundbreaking as testing an AIDS prevention vaccine in uninfected volunteers. Usually, each member presents a research study and recommends an action to the rest of committee, which then judges the request. Each protocol is reviewed in depth by four individuals on the committee, with one being the "primary reviewer." The primary reviewer then leads a discussion of the protocol for the full committee, incorporating the other reviewers' comments. The study is then open for full discussion and final vote of approval. The IRB committees are charged with ensuring that clinical research conducted at Emory meets standards aimed at protecting volunteers from unworthy investigation, from needless risk, and from misunderstanding what they have agreed to participate in. After a novel research proposal passes through an initial departmental review, the IRB may be the first group to review it, even before it is submitted for research funding. The IRB also sees the study proposal after funding has been obtained. The IRB is a patient's first and last comprehensive line of defense because much of the committee review revolves around whether patients understand what the consent form they sign says. That document spells out in simple terms the goal of the study and its risks as well as any conflict of interest a researcher may have. "We review the consent forms from the perspective of the patient and spend a lot of time discussing language changes that would make them more understandable," says Aaron Johnson, a retired hospital administrator who serves as one of several community representatives on the board. "The board is extremely conscientious. I have been surprised to see how deliberately committee members take their work and how they prepare themselves for a meeting. I am, in a word, impressed." Most of the protocols that come before an Emory IRB are approved pending changes made in the study design and/or the informed consent document. The majority are also considered low risk, which means that they are reviewed annually or if some amendment to the study is requested. Of the active and approved protocols now under way, 174 are judged to be high risk and must be reviewed by the IRB more frequently. The IRB sets a time frame to review progress or a maximum number of patients allowed in the study before a new review. Duke had a single IRB committee before its shutdown but this year has five IRB committees. Johns Hopkins had three before this summer but now has six. Emory's School of Medicine had one IRB in 1999, but now five committees consider health sciences studies, and one IRB considers requests from the rest of the campus. "Many IRBs around the country have gone from single to multiple committees to make sure there is sufficient time available to review protocols," says Rich, who serves on the Department of Health and Human Services' national human research protections advisory committee. This group serves the Office for Human Research Protections (OHRP), which replaced OPRR last year. |
 |
 |
 |
  |
mory has also almost tripled its IRB support staff since 1996 and quadrupled its budget during that time to more than $1 million. It is also considering a web-based submissions process to ease the information flow, which is now conducted in rivers of paper (20,000 pages a month) that flow through the IRB copy machine, according to Marcia Weese, IRB director.
To help researchers move their work through research hurdles, Emory opened its clinical trials office last fall under the guidance of neurologist Ray Watts. The office is expected to help investigators manage patient recruitment and research finances, among other variables, with the aim of reducing the time needed to initiate a clinical trial. Among its long-term goals is to work with the Rollins School of Public Health to help in study design and data management and analysis. A parallel office of industrial contracting has also been established to assist researchers involved in studies that are privately funded. There is also an effort to make sure research errors don't occur on the arts and sciences side of the campus. One IRB committee now considers human subject research in disciplines such as sociology, psychology, anthropology, law, history, and business. Those disciplines are generating a growing volume of research, and more and more faculty are suddenly aware they need to run their research through an IRB. If the volume of reviews in these areas continues to increase, a second committee may be created to focus on noninvasive research. The existing IRB, renamed in September as the social, humanists, and behavioral IRB, receives some interesting challenges in its mandate to protect research subjects, says its chair, Karen Hegtvedt, associate professor of sociology. A researcher studying illegal immigrants could not ask them to sign consent forms because that might constitute documentation that INS might use to ask them to leave the country. And what would informed consent mean to African villagers being observed by an Emory anthropologist? "Medical school research is fairly structured, whereas some of this research can be ambiguous and very low risk," says Hegtvedt. "But there is no question that it should be reviewed for compliance to federal guidelines." Many of these changes came about in light of Duke's shutdown and after Emory was "dinged" in a minor site visit by federal officials. In response, Emory hired consultants to ferret out deficiencies. Corrective work is ongoing, says David Blake, associate director of the Woodruff Health Sciences Center and vice president for academic affairs. The kind of mistakes seen at Penn and Hopkins "could not happen here," he now says. |
 |
"Before last year, new researchers were taught the ropes by mentors who had experience," says Victor Lampasona, former director of the human protection education program. Today clinical trial investigators must study the history, methods, and ethics of research, while associated research personnel study two of the modules. All then take an on-line, instantly graded, open-book test. By the end of November, 2,441 people had taken the test, and 2,353 had passed it, with an average score of 91. "We have heightened awareness that good research should be a learned behavior," says Lampasona. "Even seasoned researchers have told me that they learned something new." Keller, the IRB chair, is a fan of the new educational mandate. When he started chairing the committee in 1995, he worked under the assumption that "my colleagues are people of great integrity and that we should trust them. And, for the most part, I do," he says. "But the longer I am in this job, the more I realize that physicians are just not getting adequate research training in medical school." |

|
"Although there are federal and legal guidelines, many areas are subject to interpretation, as are the ethical issues," he says. "In the end we are all learning daily as we try to do our best to protect human subjects. We continually try to refine the process and do a better job as we see weaknesses in our system of review and learn from the audits of other institutions." Emory administrators are paying particular attention to the consent process -- the circumstances under which participants are asked to participate in a clinical trial. Are patients being told about the next great blood clot-dissolving drug while being wheeled in for a cardiac catheterization -- or, more appropriately, while talking face to face with a physician in a quiet room before any treatment decisions are made? Such oversight is now done on a case-by-case basis at Emory, but the OHRP has made it clear that institutions should start to monitor the process more closely, says Frank Stout, vice president for research administration. "Emory has always taken its responsibility to patients in research very seriously, and we will hire the appropriate number of ombudsmen as it takes to have confidence we are doing it right." Keller, a professor and clinical director of radiation oncology whose job as IRB chair is supposed to take only two half-days a week (the reality far exceeds that), would also like to have some system that provides oversight of data management, even laboratory assays. "Eventually we need to go further into the research process, and there is only so much auditing going on now." He says it's also necessary to review reporting of adverse events -- medical problems arising in study participants that require hospitalization or result in death. Last year, Emory researchers reported to an IRB special committee 1,421 adverse events that occurred to Emory participants in clinical trials. Such so-called adverse events can include anything that occurs to the patients under study, such as an approved deviation from a study's protocol, or even an unrelated accident, such as a fall, that requires a patient's hospitalization. Keller says most adverse events are not directly related to a drug or device being tested; in fact, only 92 (6.5%) of incidents last year were directly related to the trial the patient was enrolled in, and most of those had nothing to do with an error on the part of the researcher or health care team. Still, to completely eliminate doubt, the process needs to be streamlined, Keller says. When asked what keeps him motivated, given his huge task as guardian of human subjects, Keller doesn't hesitate: "Where else, but around this committee table, can a physician keep up to date about the latest medicine while watching the development of modern medical ethics?" For his part, Blake borrows a famous phrase from Winston Churchill to describe the immense work that has been done at Emory, with the realization that there is much more to do: "This is not the end, nor is it the beginning, but it is the end of the beginning...." For more information about institutional review boards at Emory, see www.emory.edu/IRB.
|
|
 |
 Larsen's
new study was up before a September IRB meeting. Although it took only
two weeks from submitting the study to having it heard, he had been
preparing supporting material for a long time. "We still need to streamline
the process as much as we can," says Larsen. "We need to keep the investigators
investigating and the system complying."
Larsen's
new study was up before a September IRB meeting. Although it took only
two weeks from submitting the study to having it heard, he had been
preparing supporting material for a long time. "We still need to streamline
the process as much as we can," says Larsen. "We need to keep the investigators
investigating and the system complying."
|
 |
|
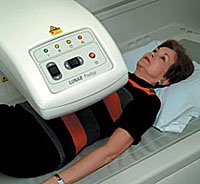
In the first clinical trial of its kind in children, scientists will learn whether putting a topical gel in children's eyes at bedtime can halt future progression of nearsightedness. Myopic children between the ages of 8 and 12, such as Joseph Zavodny of Duluth, have signed up for the year-long study at a dozen sites across the country, say pediatric opthalmalogist Arlene Drack (left), principal investigator, and study coordinator Cameile Martin (right). Although the drug Pirenzipine can't reverse myopia, it could reduce the need for stronger and stronger glasses or contact lenses as children grow older. |
 |
|
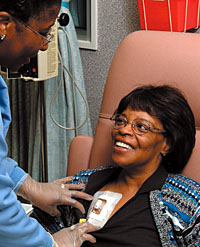
After her mastectomy in 1992, Mary Davidson volunteered for a clinical trial of the drug Tamoxifen. She was cancer-free until this past August, when it was discovered that her cancer had spread to other organs. Davidson signed up again for another multicenter trial, this one evaluating the safety and efficacy of a three-drug regimen, but was taken off that protocol after suffering serious side effects. Still, she remains undaunted. "I probably will volunteer for another trial soon," says the senior office assistant in the Winship Cancer Center. "I'm very proud to work here and have faith in our mission of finding a cure for cancer." To learn more about these studies, call Emory HealthConnection at 404-778-7777. |
 |
|
|
While the medical school has solid rules in place and an active supporting website, Emory as an institution has to guard against its own conflicts of interest, such as owning equity in a start-up company made up of campus researchers. It's a problem that universities around the country are now addressing. Sometimes the mere appearance of conflict of interest is more damaging than the facts warrant, Adkison says. "It's easy for a newspaper to run a story about Doctor X at Y university who's running a clinical trial and has a financial interest that looks inappropriate. But the article doesn't add that the conflict is being managed to the point that there is nothing amiss. That can ruin a person's reputation and instill doubt about an institution, and that's what we all guard against." In December, the Association of American Medical Colleges (AAMC) issued guidelines on financial conflicts of interest in clinical research. In the coming year, AAMC will develop guidelines to monitor institutional financial interests in human subjects research. For more information, see www.aamc.org/coitf. |

|
Copyright © Emory University, 2002. All Rights Reserved.
Send comments to the
Editors.
Web version by Jaime Henriquez.